

How Does The Cell Repair Damaged Dna?
Introduction
Over the past decade, a renewed involvement in cancer metabolism has emerged. The thought originated from the work of Otto Warburg and colleagues—first published in the 1920s—who noticed the propensity for cancer cells to consume increased quantities of glucose (1, ii). We at present understand that there is all-encompassing metabolic rewiring in cancer cells, and we are starting to decipher how cancer cells reprogram their metabolism to adapt to changes in their microenvironment and support their high metabolic needs (three–5). Cancer metabolism is a broad topic and our current agreement of how metabolic reprogramming is linked to malignant transformation has been extensively reviewed elsewhere (6–8). Therefore, in this review, we decided to focus on the connections between cell metabolism and another major aspect of cancer biology, Dna-repair/Deoxyribonucleic acid-harm pathways.
The major hallmarks, as described past Pavlova and Thompson (vii), include: deregulated glucose and amino acrid uptake, opportunistic ways of acquiring nutrients, utilize of metabolic intermediates for biomass and nicotinamide adenine dinucleotide phosphate (NADPH) synthesis, increased demand for nitrogen, and alterations in metabolite-driven gene expression and interaction with the microenvironment (seven, ix). Importantly, the wide differences in metabolic dependencies beyond cancer types, or stage of progression, also as the dynamic shifts between metabolic pathways, brand the study of cancer metabolism and the evolution of new therapies targeting these pathways very challenging.
In addition to the accepted office for cell metabolism in cancer, it is well established that Dna-repair/Deoxyribonucleic acid-damage pathways are important in cancer progression considering dysregulation leads to higher levels of genomic instability, increased mutation charge per unit, and enhanced intra-tumor heterogeneity (ten–xiii). In that location are currently three principal mechanisms through which changes in cell metabolic condition are idea to take an influence on Deoxyribonucleic acid-damage/DNA-repair pathways: chromatin remodeling, double-strand break (DSB) repair, and redox homeostasis (Figure 1). Uncovering new links betwixt these important aspects of cancer biological science might lead to the development of new targeted therapies in DNA-repair deficient cancers or even improving the efficacy of existing therapies, such as PARP inhibitors, anthracyclines, and platinum salts. In this review, we examine work seeking to uncover the links between Deoxyribonucleic acid-repair/DNA-damage and jail cell metabolism.
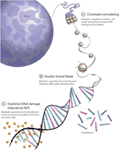
Figure i. Overview of principal links betwixt cell metabolism and DNA repair. (A) Methyl-group donors from the S-adenosylmethionine pathway and acetyl-donors from citrate bike-derived acetyl coenzyme A contribute to dynamic chromatin packaging and remodeling essential to DNA double-strand repair. (B) Metabolic intermediates derived from glucose, glutamine, and aspartate are required for de novo nucleotide synthesis. The ready availability of a puddle of nucleotides facilitates appropriate Dna repair and replication. (C) Intracellular reactive oxygen species (ROS) levels reflect a residuum betwixt generation and detoxification. A principal ROS detoxification mechanism involves reduced glutathione (GSH), determined by glutamine and cysteine availability, as well equally NADPH levels. High ROS-induced DNA damage leads to excessive burden on the Dna repair machinery.
Metabolic Status Affects Dna Folding and Repair Pathways
The first link between DNA repair and jail cell metabolism involves DNA folding and organization. Chromatin packaging and remodeling, through different histone posttranslational modifications—including acetylation, methylation, phosphorylation, and ubiquitination, likewise equally through Deoxyribonucleic acid modifications such every bit methylation—can regulate cistron expression levels past modulating the access to Dna of different protein complexes (14, fifteen). Interestingly, like mechanisms can likewise regulate admission of Deoxyribonucleic acid-repair proteins to the DNA double-helix (xvi). When repairing DSBs, the offset step involves unfolding DNA to allow access of the repair complexes to the DSB (17). There is a growing body of evidence suggesting that these mechanisms can in fact regulate the choice of Deoxyribonucleic acid-repair pathway (i.e., homologous recombination), or not-homologous terminate-joining used to repair the DSB (16). Substrates added to histones or Deoxyribonucleic acid are derived from metabolic intermediates (xviii–20) (Figure two). For example, methyl-group donors mostly come from the Due south-adenosylmethionine (SAM) pathway (18), while the sole acetyl-group donor is acetyl coenzyme A (acetyl-CoA), known for the transfer of its acetyl group to oxaloacetate to course citrate and beginning the tricarboxylic acid (TCA) cycle (21). It has been shown that expression levels of the enzyme, ATP citrate lyase, can regulate the availability of acetyl-CoA in the jail cell (22). Restricting the corporeality of acetyl-group donors can disrupt proper DNA organization and have an impact on DNA folding and DNA remodeling essential to successful Deoxyribonucleic acid DSB repair (22, 23). Acetyl-CoA tin can also be generated from acetate and CoA from the acetyl-CoA synthetase 2 enzyme. In fact, under metabolic stress weather in which oxygen and lipids are deprived, acetate-generated acetyl-CoA is enhanced and acetate-derived carbons are incorporated in lipid synthesis (24, 25). Acetate-derived acetyl-CoA has likewise been shown to exist involved in histone acetylation, suggesting that acetate availability could influence histone acetylation in cancer cells (26).
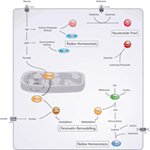
Figure two. Simplified diagram of the main metabolic pathways involved in Deoxyribonucleic acid damage/repair. Nucleotide pool: nucleotide precursor ribose-5-phosphate (R5P) is generated from the pentose-phosphate pathway (PPP). Purines and pyrimidines precursors, inosine monophosphate (IMP), and uridine monophosphate (UMP), respectively are synthesized from glutamine, and aspartate and carbamoyl phosphate, respectively. Redox homeostasis: nicotinamide adenine dinucleotide phosphate (NADPH) is generated from the PPP and the serine synthesis pathway. Glutathione (GSH) is generated from cysteine and glutamate. Chromatin remodeling: methyl-group donors (CH3) are generated from the S-adenosyl methionine (SAM) pathway and acetyl-group donors (acetyl coenzyme A) are generated from the citrate wheel or from acetate. Overabundance-1, glucose transporter one; SCL1A5, alanine, serine, cysteine-preferring transporter 2 (ASCT2); G6P, glucose 6-phosphate; GA3P, glyceraldehyde iii-phosphate; SAH, Due south-adenosyl homocysteine; xCT, cystine-glutamate antiporter; MCT, monocarboxylate transporter family.
Additionally, changes in the activity of the SAM pathway due to availability of necessary nutrients or substrates also influence DNA or histone methylation by regulating the pool of methyl-donors (27). The SAM pathway is interconnected with methionine, tetrahydrofolate (THF), 1-carbon metabolism, and choline pathways. These pathways tend to compensate for ane another; however, depletion of choline, folate, or methionine can however accept an touch on on the final SAM concentration (18). Such changes in the SAM pathway non just influence gene expression of cancer-associated genes through epigenetic modifications but can likewise touch DNA folding during DNA repair processes. The latter can besides be influenced directly by metabolic intermediates such equally fumarate. DNA-PK dependent activation of fumarase contributes to enhanced local generation of fumarate, which in plow promotes Dna repair via inhibition of KDM2B-mediated histone demethylase activity (28). Accumulation of fumarate can likewise pb to epithelial-to-mesenchymal transition and poor clinical outcome (29). Since modifications in histone and DNA are important for proper repair of Dna DSBs (30), changes in nutrient and substrate levels that disrupt these modifications are bound to affect DNA-repair pathways.
Nucleotide Levels Touch on Dna-Repair Potential of Cancer Cells
Another major mechanism upon which prison cell metabolism can regulate Deoxyribonucleic acid-repair/Deoxyribonucleic acid-impairment is through the regulation of the pool of nucleotides used for Dna replication and repair (31, 32). Many dissimilar metabolic pathways are involved in de novo nucleotide synthesis and can have an impact on the levels of intracellular nucleotides bachelor (Figure ii) (31, 33, 34). An of import precursor in the synthesis of the ribose backbone, essential for both purines and pyrimidines, comes from ribose-5-phosphate, an intermediate of the pentose-phosphate pathway (PPP) (31). Briefly, the PPP utilizes glucose-6-phosphate (G6P), an intermediate of glycolysis, which can be redirected to generate metabolic intermediates necessary for nucleotide and protein synthesis, as well every bit generation of NADPH. The PPP is a major focus in cancer metabolism, and information technology has been carefully reviewed elsewhere (31, 35, 36). The increased glucose consumption observed in cancer cells has been shown, at to the lowest degree in some cancers, to be used to fuel the PPP, where it can potentially be used to generate reducing power in the form of NADPH and nucleotide precursors (32). Additionally, other metabolic pathways are involved more specifically in purine or pyrimidine ring synthesis. The amide group of glutamine is essential in 2 steps of inosine monophosphate synthesis, an intermediate in de novo purine synthesis, and in one pace of uridine monophosphate synthesis, an intermediate in de novo pyrimidine synthesis (37, 38). In glioblastoma, it has been shown that α-ketoglutarate is diverted out of the TCA bicycle to synthesize more than glutamine through the glutamine synthetase enzyme. The resulting glutamine is then used for de novo purine synthesis (39). Furthermore, aspartate is essential for the synthesis of pyrimidines, and glycine is also essential for purine synthesis (38). Loss-of-part mutations in arginosuccinate synthase 1, which leads to reduced arginine production in the urea cycle, promotes the use of aspartate for pyrimidine synthesis (33). Aspartate is mostly synthesized from glutamate and oxaloacetate. Therefore, the availability of glutamine and/or other amino acids/metabolic substrates can influence the corporeality and ratio of nucleotides produced in a cell and might provide a regulatory machinery for Dna-repair.
Deregulated Redox Homeostasis Promotes Deoxyribonucleic acid Damage
The metabolic regulation of reactive oxygen species (ROS) levels is the tertiary and concluding link that we address here between Dna-repair/Deoxyribonucleic acid-damage and prison cell metabolism. High ROS levels affect many aspects of tumor biology and, hither, we focus on their role in inducing Dna damage and genomic instability. Most of the Dna lesions that are formed past ROS-induced impairment are unmarried strand breaks (SSBs) that can be repaired through nucleotide or base of operations excision repair (NER/BER) (40). Still, these SSBs can lead to stalling of the replication fork or error in replication, ultimately leading to DSBs (40). The accumulation of oxidative Dna impairment increases the burden on the Deoxyribonucleic acid-repair machinery. Tight regulation of cellular redox stress is essential, since loftier ROS levels can atomic number 82 to oxidative stress and oxidative damage of proteins, Dna, and lipids, while a certain level of ROS is essential for activating signaling pathways involved in multiple biological processes (41–43). Cells accept evolved a number of ways to residuum ROS levels (Effigy 2). Glutathione (GSH) is one of the major ROS-scavenging molecules (44), and it occurs in two versions; the reduced class, sulfhydryl GSH, and the oxidized, glutathione disulfide (GSSG). The enzyme GSH peroxidase catalyzes the reduction of H2Otwo to water and lipid hydroperoxides to their corresponding lipid alcohols and in turn oxidizes GSH to GSSG. The contrary reaction is catalyzed by the enzyme glutathione reductase, also known as glutathione-disulfide reductase using the reducing potential of NADPH (45).
Glutathione is synthesized in the cytoplasm from cysteine and glutamate, and cysteine availability is the rate-limiting step of the synthesis reaction (46, 47). Cysteine tin be synthesized from serine through the transsulfuration pathway (47), or information technology tin can exist transported into the cells as cystine in substitution for glutamate using the cystine-glutamate antiporter xCT (39). Interestingly, mTORC2 can phosphorylate and inhibit the xCT antiporter, therefore providing a mechanism to regulate intracellular cysteine levels (48). Furthermore, it has been shown that, in some cancer types, growing cells in DMEM containing cystine leads to xCT dependent glutamine dependency in vitro, providing testify for context dependent changes in metabolism (5). The main source of NADPH comes from the PPP and a meaning amount as well comes from the serine synthesis pathway through the THF pathway. Both harness the free energy from the catabolism of metabolic intermediates to generate NADPH (31, 47). Interestingly, an important regulator of ROS levels is the transcription factor nuclear factor E2-related factor 2 (NRF2) that has been shown to straight regulate the key serine synthesis enzymes (PHGDH, PSAT1, ATF4) (49). This provides a potential mechanism through which NRF2 can regulate ROS levels by influencing NADPH and GSH levels. Importantly, the DNA-repair protein, BRCA1, regulates NRF2, providing an additional link between DNA repair pathways and ROS levels (l). Overall, in that location are all the same many open questions with regards to how cancer cells can rewire their metabolism based on different nutrient availability, but there is growing show to suggest that maintaining cellular redox homeostasis plays a major office on Deoxyribonucleic acid-damage/DNA-repair pathways.
DNA Damage Response (DDR) Triggers Metabolic Rewiring
While DNA-repair pathways can be influenced past cellular metabolic condition and food availability in the tumor microenvironment, aggregating of Dna impairment due to extrinsic and intrinsic genotoxic stress, or scarce DNA repair can also crusade sharp rewiring of jail cell metabolism. Cells have evolved the DDR pathway (DDR) to monitor this genotoxic stress and maintain authentic transmission of genetic information to subsequent generations. The DDR can, therefore, halt cell-bicycle progression, induce Deoxyribonucleic acid-repair mechanisms, or trigger programmed cell death when DNA damage is irreparable (51). Clutter telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinases are 2 fundamental enzymes in the recognition of Deoxyribonucleic acid harm and implementation of the DDR (51, 52). Upon activation by Dna-harm, ATM and ATR generate a 2nd wave of phosphorylation that impacts many downstream effector proteins. An of import aspect of DDR, driven past ATM and ATR activation, is the consecration of metabolic rewiring to promote the resolution of genotoxic stress. ATM has been shown to activate the PPP through induction of the charge per unit-limiting enzyme glucose-6-phosphate dehydrogenase (G6PD), to support the synthesis of reducing ability in the form of NADPH, and generate ribose-5-phosphate for nucleotide synthesis (53, 54).
Increased carbon flux to the PPP from glucose derivatives is driven not only by increased glucose consumption ofttimes observed in cancer cells (55, 56) but also through ROS-mediated inactivation of many glycolytic enzymes, notably PKM2 that promotes flux into the oxidative arm of the PPP too every bit serine biosynthesis (57). Many studies have shown increased glucose dependency post-obit DDR (58), and these data demonstrate the challenge posed to cancer cells under genotoxic stress. They need to contain ROS levels, enhance nucleotide synthesis to repair DNA damage, and inhibit cell-bike progression until resolution. To this terminate, SIRT4 activation upon DDR has been shown to inhibit glutamine consumption in HepG2, HeLa, HEK293T cells, and in lung tissue (58). The reduced glutamine consumption or, more than specifically, reduced anaplerosis of glutamine into the TCA bicycle, confers improved cell survival compared to SIRT4 knockout cells that cannot actuate this response (58). While this study suggests that DDR undoubtedly causes changes in metabolism, conflict in the literature indicates that the nature of these changes is context- and cell type-dependent. For example, the concept that cells undergoing genotoxic stress demand to synthesize more nucleotides to repair the DNA damage contradicts the reduced glutamine consumption. As mentioned previously, the amide group of glutamine is essential for purine and pyrimidine de novo synthesis. Other interesting points to notation are that in non-cancerous tissues/cells, there are increases in fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) in response to acute or chronic genotoxic stress (59). Increased FAO/OXPHOS is driven by AMP kinase (AMPK) activation resulting from the depletion of ATP. In the context of that written report, cells are depleted of ATP because of the utilization of NAD+ by the Deoxyribonucleic acid repair enzyme PARP-1. PARP-i generates PAR chains from NAD+ and PARylates Deoxyribonucleic acid to induce repair. This leads to depletion of NAD+, reduction of ATP synthesis, and activation of AMPK (59). This study further supports that Deoxyribonucleic acid impairment impacts cell metabolism. However, given the current literature, it is still unclear how a cell's nutrient availability can dictate the nature of its metabolic rewiring.
Metabolic Changes Driven by DDR Factor Mutations
The p53 poly peptide encoded by the TP53 cistron is some other key thespian in DDR through its role in DNA repair and cell-cycle regulation (lx–62). While p53 levels are kept low under normal conditions, upon DNA damage or other cellular stress, p53 is activated by phosphorylation events that prevent its deposition (sixty). Wild-type p53 activation regulates cell-cycle entry past transcriptionally repressing cyclin B and CDC25B, and information technology also plays an important role in Deoxyribonucleic acid-repair. More specifically, it determines whether or non to repair the DNA or trigger senescence or apoptosis. p53 also plays many modulatory roles in the metabolic rewiring of cells. Among these, is its regulation of glycolysis through transcriptional activation of the TP53-induced glycolysis and apoptosis regulator (TIGAR) protein (63). TIGAR inhibits glycolysis by dephosphorylating fructose-2,half dozen-biphosphate (F2,6B) leading to degradation of F2,6P and inhibition of phosphofructo kinase 1, an enzyme integral to glycolysis. p53 has too been shown to inhibit other enzymes of glycolysis as well every bit straight repress the expression of glucose transporters, GLUT1 and GLUT4 (61, 64). 1 might surmise that this impediment in glycolysis potentially leads to a surplus in glycolytic intermediates, leading to redirection toward the PPP. Nevertheless, p53 has also been shown to inhibit the flux of glucose to the PPP by directly binding to G6PD and preventing its active dimer formation (61).
In improver, p53 has been shown to transcriptionally regulate proteins involved in the electron transport chain and mitochondrial stability, therefore, providing a mechanism to regulate OXPHOS (61, 65). Together, these data suggest that the role of p53 in metabolic regulation is context and tissue dependent. Importantly, TP53 is one of the most mutated genes in cancer and many proceeds-of-function mutations have been described, leading to unlike changes in p53 depending on the mutation and the context. For example, mutant p533KR cannot induce cell-cycle-arrest, senescence, or apoptosis, but information technology can still regulate metabolic target genes resulting in decreased ROS levels and reduced glycolytic flux (61, 66). Therefore, mutations in DDR or Deoxyribonucleic acid-repair genes may result from the application of selective pressures established past the metabolic niches of cancer cells.
Other genes involved in Dna-repair are besides commonly mutated in cancer (13). Some of the most well-known DNA-repair genes associated with cancer are BRCA1 and BRCA2 but mutations in other genes such as ATM, ATR, and CDK12 [for a more extensive list, see Ref. (xiii)] as well lead to Deoxyribonucleic acid-repair scarce cancers (xiii, 67–70). Some cancers with a similar phenotype to Deoxyribonucleic acid-repair mutant cancer do not harbor any mutations in those pathways. Together, these DNA-repair deficient cancers have been described every bit having a "BRCAness" phenotype (13, 71). Some metabolic changes have been described in the context of BRCAone or BRCA2 mutations (fifty, 72–74); withal, information technology is nevertheless unclear whether these are due to their DNA-repair function or some culling office. Since connections have been drawn between metabolism and DNA-repair, it seems probable that the "BRCAness" phenotype and BRCA1/2 mutations would be associated with changes in metabolism. Once more, as with nearly other mutations found in cancer, the metabolic changes associated with these DNA repair defects might reflect the context and tissue of origin.
Conclusion
Historically, cancer has been thought of as a genetic disease driven by the aggregating of multiple mutation "hits" (75–77). However, in recent years, this paradigm has begun to shift, and cancer is often regarded as a "metabolic disease" (78, 79), which is influenced by complex interactions between the tumor and its microenvironment. Therefore, when examining data suggesting that mutations atomic number 82 to the rewiring of cancer metabolism, in fact, it may be more pertinent to question whether these mutations arise in the start place from selective pressures practical by extrinsic metabolic factors in the tumor microenvironment. In a noteworthy case, while KRAS-mutant cancers exhibit resistance to low-glucose growth atmospheric condition, glucose deprivation has been shown to drive the accumulation of novel KRAS mutations in KRAS wild-blazon cancers (fourscore). This indicates that extrinsic factors, such as the availability of essential nutrients, directly influence the fate of resulting genetic alterations that confer growth and survival advantages of cancer cells in their given microenvironment.
With regards to mutations in DNA-repair genes, it is possible that the highly dynamic and fluctuating conditions encountered in the tumor microenvironment provide an evolutionary force per unit area to select for cancer cells that have heightened genomic instability and are more proficient at adapting to these changes as a result. Consistently, it has been shown that tumors with increased heterogeneity are less responsive to therapy and behave more aggressively than tumors arising from single clonal populations (81–83). As we have discussed, a multitude of extrinsic metabolic factors affects Deoxyribonucleic acid repair and, subsequently, genomic stability (Figures one and ii). Therefore, when considering time to come strategies to refine existing or develop novel cancer therapies, information technology volition exist essential at all stages of enquiry and drug design to have account of the dynamic interplay between microenvironment and metabolic factors that are proving to influence handling efficacy and then substantially.
Author Contributions
MOT and GP wrote the manuscript. MOT and NP designed the figure. NP drew the figure and contributed to the revision of the manuscript, and bibliography.
Conflict of Interest Statement
The authors declare that the enquiry was conducted in the absence of whatsoever commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MOT and NP are supported by Cancer Research United kingdom of great britain and northern ireland Ph.D. studentships. Work in the GP lab is supported by the Institute of Cancer Research and a Cancer Inquiry UK One thousand Challenge accolade (C59824/A25044).
References
one. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi:x.1085/jgp.8.vi.519
CrossRef Full Text | Google Scholar
5. Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife (2017) 6:e27713. doi:10.7554/eLife.27713
PubMed Abstruse | CrossRef Full Text | Google Scholar
8. Martinez-Outschoorn UE, Peiris-Pages Thousand, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol (2017) 14(ii):113. doi:10.1038/nrclinonc.2017.1
CrossRef Full Text | Google Scholar
11. Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, et al. Genomic landscape of Deoxyribonucleic acid repair genes in cancer. Oncotarget (2016) 7(17):23312–21. doi:10.18632/oncotarget.8196
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Gavande NS, VanderVere-Carozza PS, Hinshaw Hard disk drive, Jalal SI, Sears CR, Pawelczak KS, et al. Deoxyribonucleic acid repair targeted therapy: the past or time to come of cancer treatment? Pharmacol Ther (2016) 160:65–83. doi:10.1016/j.pharmthera.2016.02.003
PubMed Abstract | CrossRef Total Text | Google Scholar
fifteen. Miller JL, Grant PA. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell Biochem (2013) 61:289–317. doi:x.1007/978-94-007-4525-4_13
PubMed Abstract | CrossRef Total Text | Google Scholar
18. Niculescu Doc, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr (2002) 132(eight Suppl):2333S–5S.
PubMed Abstract | Google Scholar
19. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a primal metabolite and second messenger. Cell Metab (2015) 21(6):805–21. doi:10.1016/j.cmet.2015.05.014
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Jail cell Biol (2014) fifteen(8):536–50. doi:10.1038/nrm3841
PubMed Abstract | CrossRef Total Text | Google Scholar
22. Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J, et al. Nuclear acetyl-CoA product by ACLY promotes homologous recombination. Mol Cell (2017) 67(ii):252–65.e256. doi:10.1016/j.molcel.2017.06.008
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab (2014) 2:23. doi:10.1186/2049-3002-2-23
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Jail cell (2015) 27(1):57–71. doi:10.1016/j.ccell.2014.12.002
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Mews P, Donahue K, Drake AM, Luczak V, Abel T, Berger SL. Acetyl-CoA synthetase regulates histone acetylation and hippocampal retentivity. Nature (2017) 546(7658):381–6. doi:10.1038/nature22405
PubMed Abstract | CrossRef Total Text | Google Scholar
27. Mehrmohamadi 1000, Mentch LK, Clark AG, Locasale JW. Integrative modelling of neoplasm Deoxyribonucleic acid methylation quantifies the contribution of metabolism. Nat Commun (2016) vii:13666. doi:x.1038/ncomms13666
PubMed Abstruse | CrossRef Total Text | Google Scholar
28. Jiang Y, Qian X, Shen J, Wang Y, Li Ten, Liu R, et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol (2015) 17(nine):1158–68. doi:10.1038/ncb3209
PubMed Abstruse | CrossRef Total Text | Google Scholar
29. Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa As, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature (2016) 537(7621):544–7. doi:10.1038/nature19353
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Russo G, Landi R, Pezone A, Morano A, Zuchegna C, Romano A, et al. Dna damage and repair modify DNA methylation and chromatin domain of the targeted locus: mechanism of allele methylation polymorphism. Sci Rep (2016) half-dozen:33222. doi:10.1038/srep33222
PubMed Abstruse | CrossRef Full Text | Google Scholar
33. Rabinovich S, Adler Fifty, Yizhak Grand, Sarver A, Silberman A, Agron South, et al. Diversion of aspartate in ASS1-scarce tumours fosters de novo pyrimidine synthesis. Nature (2015) 527(7578):379–83. doi:10.1038/nature15529
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Kim J, Hu Z, Cai Fifty, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and Dna synthesis in KRAS/LKB1-mutant lung cancer cells. Nature (2017) 546(7656):168–72. doi:10.1038/nature22359
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5(8):592–602. doi:x.1007/s13238-014-0082-eight
CrossRef Full Text | Google Scholar
37. Cory JG, Cory AH. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: asparaginase treatment in childhood astute lymphoblastic leukemia. In Vivo (2006) xx(5):587–9.
PubMed Abstract | Google Scholar
39. Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Jail cell Biol (2015) 17(12):1556–68. doi:10.1038/ncb3272
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Alleman RJ, Katunga LA, Nelson MA, Brownish DA, Anderson EJ. The "Goldilocks Zone" from a redox perspective-adaptive vs. deleterious responses to oxidative stress in striated musculus. Front Physiol (2014) 5:358. doi:x.3389/fphys.2014.00358
CrossRef Total Text | Google Scholar
44. Shanware NP, Mullen AR, DeBerardinis RJ, Abraham RT. Glutamine: pleiotropic roles in tumor growth and stress resistance. J Mol Med (Berl) (2011) 89(3):229–36. doi:10.1007/s00109-011-0731-9
PubMed Abstruse | CrossRef Full Text | Google Scholar
46. Hultberg B, Hultberg M. Loftier glutathione turnover in human cell lines revealed by acivicin inhibition of gamma-glutamyltranspeptidase and the effects of thiol-reactive metals during acivicin inhibition. Clin Chim Acta (2004) 349(1–2):45–52. doi:10.1016/j.cccn.2004.05.024
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Yang Grand, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer (2016) 16(10):650–62. doi:10.1038/nrc.2016.81
CrossRef Full Text | Google Scholar
48. Gu Y, Albuquerque CP, Braas D, Zhang West, Villa GR, Bi J, et al. mTORC2 regulates amino acrid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell (2017) 67(1):128–38.e127. doi:x.1016/j.molcel.2017.05.030
PubMed Abstruse | CrossRef Full Text | Google Scholar
49. DeNicola GM, Chen PH, Mullarky Due east, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small-scale cell lung cancer. Nat Genet (2015) 47(12):1475–81. doi:10.1038/ng.3421
PubMed Abstract | CrossRef Total Text | Google Scholar
50. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and jail cell survival. J Exp Med (2013) 210(viii):1529–44. doi:10.1084/jem.20121337
PubMed Abstract | CrossRef Full Text | Google Scholar
52. Mirzayans R, Severin D, Murray D. Relationship betwixt DNA double-strand break rejoining and cell survival later on exposure to ionizing radiation in human fibroblast strains with differing ATM/p53 status: implications for evaluation of clinical radiosensitivity. Int J Radiat Oncol Biol Phys (2006) 66(5):1498–505. doi:ten.1016/j.ijrobp.2006.08.064
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence force and Dna repair. EMBO J (2011) 30(3):546–55. doi:ten.1038/emboj.2010.330
PubMed Abstract | CrossRef Total Text | Google Scholar
54. Aird KM, Worth AJ, Snyder NW, Lee JV, Sivanand S, Liu Q, et al. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep (2015) eleven(6):893–901. doi:10.1016/j.celrep.2015.04.014
PubMed Abstract | CrossRef Full Text | Google Scholar
57. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen One thousand, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Scientific discipline (2011) 334(6060):1278–83. doi:10.1126/scientific discipline.1211485
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell (2013) 23(4):450–63. doi:x.1016/j.ccr.2013.02.024
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Caryatid LE, Vose SC, Stanya K, Gathungu RM, Marur VR, Longchamp A, et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech Dis (2016) ii:16022. doi:10.1038/npjamd.2016.22
PubMed Abstract | CrossRef Full Text | Google Scholar
64. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli Due east. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 factor expression. Cancer Res (2004) 64(7):2627–33. doi:x.1158/0008-5472.Tin can-03-0846
PubMed Abstract | CrossRef Full Text | Google Scholar
66. Li T, Kon N, Jiang L, Tan Chiliad, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-wheel arrest, apoptosis, and senescence. Prison cell (2012) 149(6):1269–83. doi:ten.1016/j.jail cell.2012.04.026
PubMed Abstruse | CrossRef Full Text | Google Scholar
67. Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong Northward, et al. Germline BRCA1 mutations and a basal epithelial phenotype in chest cancer. J Natl Cancer Inst (2003) 95(nineteen):1482–five. doi:10.1093/jnci/djg050
PubMed Abstract | CrossRef Full Text | Google Scholar
69. Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, Gonzalez-Angulo AM, et al. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative chest cancer. J Clin Oncol (2008) 26(26):4282–viii. doi:x.1200/JCO.2008.sixteen.6231
PubMed Abstract | CrossRef Full Text | Google Scholar
70. O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: chest/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis (2010) 31(half-dozen):961–vii. doi:ten.1093/carcin/bgq069
PubMed Abstract | CrossRef Full Text | Google Scholar
72. Privat Thousand, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, et al. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS One (2014) 9(7):e102438. doi:10.1371/journal.pone.0102438
PubMed Abstract | CrossRef Total Text | Google Scholar
73. Rytelewski M, Tong JG, Buensuceso A, Leong HS, Maleki Vareki S, Figueredo R, et al. BRCA2 inhibition enhances cisplatin-mediated alterations in tumor prison cell proliferation, metabolism, and metastasis. Mol Oncol (2014) viii(8):1429–xl. doi:x.1016/j.molonc.2014.05.017
PubMed Abstract | CrossRef Total Text | Google Scholar
74. Cuyas E, Fernandez-Arroyo S, Alarcon T, Lupu R, Joven J, Menendez JA. Germline BRCA1 mutation reprograms breast epithelial prison cell metabolism towards mitochondrial-dependent biosynthesis: evidence for metformin-based "starvation" strategies in BRCA1 carriers. Oncotarget (2016) 7(33):52974–92. doi:ten.18632/oncotarget.9732
CrossRef Full Text | Google Scholar
76. Ashley DJ. The two "hit" and multiple "hit" theories of carcinogenesis. Br J Cancer (1969) 23(2):313–28. doi:10.1038/bjc.1969.41
CrossRef Total Text | Google Scholar
79. Seyfried TN, Flores RE, Poff AM, D'Agostino DP. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis (2014) 35(three):515–27. doi:10.1093/carcin/bgt480
PubMed Abstract | CrossRef Total Text | Google Scholar
lxxx. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science (2009) 325(5947):1555–9. doi:x.1126/scientific discipline.1174229
PubMed Abstruse | CrossRef Full Text | Google Scholar
82. Jogi A, Vaapil M, Johansson M, Pahlman S. Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Ups J Med Sci (2012) 117(2):217–24. doi:10.3109/03009734.2012.659294
PubMed Abstruse | CrossRef Full Text | Google Scholar

How Does The Cell Repair Damaged Dna?,
Source: https://www.frontiersin.org/articles/10.3389/fonc.2018.00015/full
Posted by: olivenonvize.blogspot.com
0 Response to "How Does The Cell Repair Damaged Dna?"
Post a Comment